Методические вопросы геологии. О грубых ошибках в решении геологических задач
УДК 551.1/4. + 550.8.01
МЕТОДИЧЕСКИЕ ВОПРОСЫ ГЕОЛОГИИ. О ГРУБЫХ ОШИБКАХ В РЕШЕНИИ ГЕОЛОГИЧЕСКИХ ЗАДАЧ.
В.П.Макаров
Российский государственный геологоразведочный университет, г. Москва.
Введение
В работе [1] проанализированы основные методологические вопросы научного геологического познания и намечены некоторые проблемы, искажающие решения геологических задач. Главный вывод заключался в том, что практически все кандидатские и докторские диссертации по отрасли "Геолого-минералогические науки" не являются отражением решения научных задач, а суть производственные отчёты, выполненные под покрывалом «решения научных задач». Они решают сугубо технологические проблемы, не имеющие прямого отношения к науке.
Известно [27], что каким бы ни было научное исследование, при его проведении всегда выделяются этапы: прямая и обратная задачи. Геология - это решение обратных задач. Наличие решений прямых задач говорит только о том, что решение обратной задачи возможно и существует, но его нужно искать самостоятельно, хотя и опираясь на решение прямой задачи.
Решение геологических задач, прежде всего, исходит из результатов геологического наблюдения, которое представляет собой один из методов решения обратной задачи геологии [27]. Но геологическое наблюдение устанавливает только гипотетические связи между геологическими объектами, составляющие основу гипотезы. К тому же, как бы тщательно не проводились геологические наблюдения, их геологическая интерпретация, т.е. сам факт решения геологической задачи, всегда поверхностна и примитивна, как любое эмпирическое действие. Ясно, что для превращения гипотетического и вероятного знания в объективную реальность необходимо аккуратно использовать существующие физическое, физико-химическое и пр. знания.
Ниже более детально рассмотрены особенности решения некоторых геологических задач.
При анализе использованы следующие критерии оценки качества:
1). "Наука" – это систематизированный результат научной деятельности, в процессе осуществления которой происходит поиск и изучение объективного знания. Объективно существующие явления и связи между ними объединяются понятием "истина". Поиск и изучение истины – это два этапа осуществления научной деятельности, синонимом которой является "научное исследование" [1].
2). Согласно В.И. Ленину «… грамотность определяется не тем, сколько «измов» познал товарищ, а тем, как эти «измы» реализуются на практике» [2].
3). По Ф. Энгельсу: «десять паровозов доказывают истинность термодинамики не более, чем один паровоз» [3].
1. Геохронология
Методологические ошибки в решении геохронологических задач ранее рассмотрены в работе [4] на примере преимущественно U- Pb и Pb- Pb систем. Основной вывод заключался в том, что все уравнения [5], использованные с этой целью, не выведены, а выдуманы; понятие о «дискондартности» выдуманно и предназначено для сокрытия того факта, что неверно вводятся поправки на «обыкновенный свинец». Пересказывать статью не будем, а повторим только один важный вывод, относимый к интерпретации взаимоотношения конкордии и изохроны в системе координат 207Pb/235U – 206Pb/238U. Для анализа производится сравнение с прямой линией, описываемой уравнением 207С= а206С + А [25]. Соотношения точек в обеих системах координат показаны на рис.№1. Если поправки на обыкновенный свинец введены не верно, то точка на конкордии растягивается в прямую D*К*А- изохрону (рис. 1Б), названную псевдоизохроной. Ее угловой коэффициент прямого отношения к возрасту уже не имеет. Одно из свойств: при перемещении текущей точки Аk в сторону осей координат в ней уменьшается количество Pbo и увеличивается количество Pbp. На этом основан способ определения времени проявления стадий минералообразования. Так по [5] первая точка пересечения псевдоизохроны и конкордии «отвечает значению возраста их образования», тогда как вторая - «верхний предел времени дифференциации U и Pb в земной коре» [5, стр.24]. Есть и другая интерпретация этих точек: если вторая точка отвечает времени становления
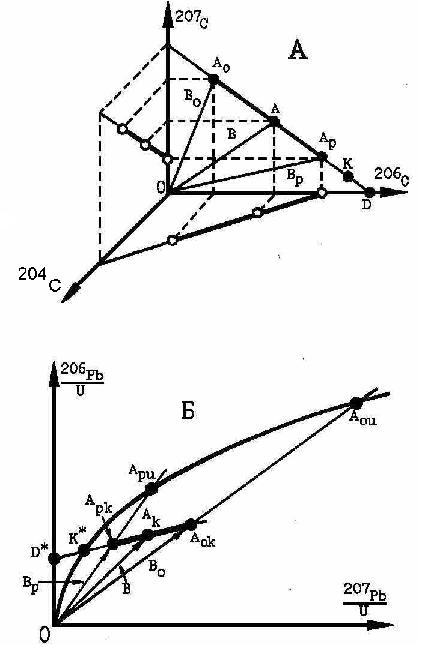
Рис.№ 1. Поведение точек в геохронологических системах координат. nbsp;
породы, то первая - времени наложенной минерализации. Ясно, что это – разночтения одного и того же явления. Обе интерпретации не соответствуют свойству псевдоизохроны.
Отметим главную и принципиальную ошибку: по умолчанию полагается, что точки пересечения конкордии и псевдоизохроны, например, точка К* (рис.1Б), имеют реальный физический смысл. Но доказательство этому не приводится, оно не существует. Поскольку этим точкам придается большое значение, мы повторим доказательство отсутствия у этой точки физического смысла. Для доказательства приводится рис. №1А. Между системами рис.№1А и рис.№1Б существует жесткая связь.
Действительно на рис. №1А точка А- текущая, ей соответствуют координаты 206СА и 207СА. Тогда угловой коэффициент SA прямой ОА равен SA = 207СА/206СА. На рис.№1Б этой точке соответствует точка Ак с координатами (206Pb/238U)Ак и (207Pb/235U)Ак. Для отрезка ОАк угловой коэффициент SAк = (206Pb/238U)Ак/(207Pb/235U)Ак. Проведя соответствующие преобразования выражения для SAк, в итоге получим соотношение
(SA× R ×SAк)= 1.
Оно важно тем, что положение луча на рис. №1А с его помощью можно перенести на рис. №1Б и получить на рис. №1Б образ любой прямой из рис. №1А. Образом отрезка АоАр является отрезок АокАрк. Угол АоОАр преобразуется в угол АокОАрк. Следовательно, образ любого отрезка АоАр будет располагаться только в угле АокОАрк и концевые точки отрезка АокАрк будут скользить по сторонам угла АокОАрк. На рис. №1А точка Ар по построению отражает точное соотношение между радиогенными компонентами. Она соответствует величине 204С = 0. Положение же точки K соответствует условию 204С < 0, что не возможно. Поэтому она не имеет физического смысла. Это означает, что и любая точка на интервале АрD также не имеет физического смысла.
На рис. №1Б образом точки D является точка D*, точки Ар – точка Арк, интервала АрD – интервал АркD*. Следовательно, любая точка на этом интер-вале АркD* не имеет физического смысла. Образом точки K является точка К*, следовательно и эта точка реального физического смысла не имеет, что и следовало доказать. Следовательно, выводы, полученные на основе использования системы координат 207Pb/235U – 206Pb/238U, не верны.
2. Решения обратной задачи термодинамики – геобаротермометрия.
Одним из важных методов изучения геологических процессов является применение различных геохимических и изотопных геотермометров (далее термодинамических геотермометров). Существенный вклад в развитие направления внёс Л.Л. Перчук, предложивший теоретическую концепцию этого направления, названную им «Теорией фазового соответствия» [6] и получившую широкое распространение в геологической практике. Он использовал представления о «химическом равновесии» между минералами, понятием в его изложении достаточно расплывчатым, не доказанным, а потому спорным. Трудно говорить о химическом равновесии, например, между природными биотитом (Bio) и калиевым полевым шпатом (Kf) в одной и той же породе, образованными при разных температурах, а значит и в разное время (первый - при Т ≈ 700оС, второй - при Т ≈ 400 - 500оС) [14]. То же можно сказать и о паре - биотит-мусковит (Тобр(Mus) ≈400oC). К тому же и Л.Л. Перчук в более ранних работах, и другие исследователи (Н.Н. Фонарёв и др.) в экспериментах показали, что это равновесие устанавливается в лучшем случае в 10%.
Практика показала, что технология их применения не соответствует теоретической схеме. Действительно, используя представления о реакциях обмена, на которых базируются геотермометры, эту реакцию в общем виде можно записать следующим образом:
А* + Б = А + Б*, (1)
где (*) отражает обогащение одного минерала некоторым элементом либо изотопом элемента. Далее привлекаются термодинамические изыски. Мы их не касаемся, хотя и здесь имеются ошибки, в частности, в учёте представлений о размерности величин [7]. Например, не корректно используется выражение
![]() (2)
(2)
где A = -ΔHo – энтальпия, B = ΔSo – энтропия реакции. K – коэффициент распределения. Показано [30], что это выражение содержит две ошибки: а) величина К не является безразмерной величиной, как это по умолчанию предполагается; поэтому выражение lnK не имеет смысла; б) это не уравнение, а равенство с тремя переменными вида, применённое к локальной точке i,
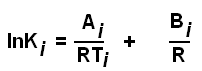
поэтому величины lnK, А и В зависят от температуры и давления.
Главное свойство уравнения (1) заключается в том (необходимый признак), что оно отражает наличие термодинамического (геохимического или изотопного) равновесия между минералами. Именно оно позволяет использовать теоретическую физико-химическую базу, а значит для любой конкретной природной пары, выбранной в качестве геотермометра, должно устанавливаться наличие этого равновесия. При практическом использовании геотермометра должны соблюдаться ещё и других условия (достаточные признаки), способствующие протеканию этих видов обмена: а) минералы должны выделяться одновременно; б) температуры образования (Тобр) минералов должны быть равными; в) минералы должны контактировать друг с другом, хотя в этом случае возможно проявление диффузионных эффектов. В крайнем случае, они должны находиться близко друг от друга; г) между минералами должен протекать изотопный или ионный обмен. Эти признаки характеризует технологические условия использования геотермометров (ГТ).
Однако в практических исследованиях эти условия никогда не проверяются. Так Л.Л. Перчук [8], обосновывая гранат-биотитовый (Grn-Bio) ГТ, никогда не определял наличие между минералами термодинамического равновесия в реальных условиях. Более того в работе [9] он производит сопоставление термопар Grt-Bio и Grt-Cor (кордиерит). Мало того, что он не установил наличие равновесия между реальными природными минералами каждой пары, но ещё по умолчанию предположил, что Grt может быть одновременно равновесен сразу двум минералами (Bio← Grt →Cor). Но теория обмена в бытность Л.Л. Перчука не рассматривала подобные равновесия.
Авторитарный характер руководства геологическими исследованиями СССР и России и вследствие этого жесточайшая цензура в геологической печати способствовали широкому распространению в геологической практике именно подобных исследований. Так В.А. Курепин [10] подобным образом сравнивает Grt - Cor и Grt – Bio – геотермометры, В.М. Венедиктов и др. [12] проводит согласование геотермометров Grt-Cpx, Grt-Opx, Opx-Cpx, Grt-Bio, причём здесь Grt, оказывается, должен быть равновесен сразу трём минералам – клинопироксену Cpx, ортопироксену Opx и биотиту Bio.
В предыдущие годы температуры образования мономинералов только оценивались по некоторым признакам. В последующем в связи с проведением анализа газово-жидких (ГЖВ) и расплавных включений (РВ) [13], а также применением изотопных методов [14] появилась возможность определения Тобр конкретного минерала. Они показали, что реальные Тобр не соответствуют принятым значениям. Результаты этих исследований выявили не только несовершенство методики Л.Л. Перчука, поскольку он полностью игнорировал Тобр конкретных минералов и состояние равновесия между минералами, но и надуманность практически всех предлагаемых им ГТ. Следовательно, оценки вида, например, Тобр = 570оС для анартозитов [15, с.175] или Тобр =360оС для пелитовых сланцев [там же, с.172] являются блефоми. Не состоятельны также геотермометры “хлорит-биотитовый”, “хлорит – гранатовый”, “Mus – Bio” и им подобные, поскольку все минералы этих пар образованы при разных Тобр, а потому и в разное время (однако обратное не корректно).
С целью объяснения различий данных, полученных разными ГТ, вместо детального анализа причин этого явления стали использовать материалы по концентрациям сосуществующих элементов, например, фтора и др. [13]. Эта методика оказалась заразительной среди российских, да и зарубежных, исследователей. Но попытки учета этого явления приводят к громоздким уравнениям расчетов, для решения которых требовалось применение ЭВМ при полном отсутствии критериев оценки достоверности получаемых при этом результатов. Поэтому в настоящее время намечается даже отказ от введения таких поправок [18] (а также B. Wood,1974; T.Morry, 1975).
В работе [6] с гордостью говорится, что в настоящее время "Теория равновесий минералов переменного состава - теория фазового соответствия - стала обычным курсом многих геологических вузов, а основанная на ней термобарометрия теперь считается в геологии единственным точным методом определения РТ- параметров формирования кристаллических горных пород. Число откалиброванных термометров и барометров (имеется в виду геохимических) очень велико. Благодаря систематическому повышению их точности можно судить не только о РТ- параметрах формирования горных пород, но и об их эволюции, то есть об их погружении на большие глубины и/или подъеме к поверхности Земли".
Проведённый выше анализ, а также анализ этой рекламки показывает, что в практическом исполнении "теория" фазового соответствия полна грубых ошибок, которые сводят на нет громадную работу по изучению природы и эволюции геологических объектов и явлений. Созданных ГТ весьма много, но как говорится "на дармовщинку и уксус сладкий": примитивизм в подходе к созданию и использованию ГТ позволяет шлёпать их как оладьи на сковородке, знай только, лей тесто и всё будет хорошо.
Полностью такая же ситуация и в изотопной ГТ-ии. Основателями практической изотопной геотермометрии были американцы J. Bigeleisen и Y. Bottinga. Вместе с этим, не проанализировав уравнения изотопных геотермометров (ИГТ), они стали и отцами грубейших ошибок в их практическом использовании, которые расползлись по всему миру и были подхвачены нашими любителями «клубнички» (В.А. Гриненко, Е.Н. Донцова, В.И. Устинов, В.Б. Поляков и др.). Примерами неверных ИГТ являются Qw-Mt; Qw-Mus [19, 20], Py-Gn [21] и др. Авторы не только не доказывали наличие между ними изотопного равновесия, но и включали в состав пар минералы, образованные при разных температурах (кварц-магнетит; кварц - мусковит, пирит - галенит и пр.). Наши последыши, абсолютизировав эту методику, пошли ещё дальше. Вместо того, чтобы внимательной проанализировать источники различий, показываемые разными ИГТ, стали выдумывать механизмы, объясняющие эти различия. В этом преуспел, в частности, В.И. Устинов [26], который «предложил» учитывать скорости реакций обмена изотопами. Хотя сама по себе эти идея заслуживает внимания, реализация её просто некорректна.
«В последнее десятилетие прошлого столетия и в начале нынешнего были подвергнуты ревизии прежние калибровки геотермометров, основанные на работах 60-х и 70-х годов. При этом использование новых методов, как экспериментальных, так и теоретических, позволило существенно уточнить температурные зависимости равновесных изотопных факторов для карбонатов, магнетита, графита, минералов, наиболее важных с точки зрения применения стабильных изотопов в геохимии щелочных пород.»[28]. Практика показывает, что эта реклама - просто фикция, попытка прикрыть высокопарными словами процветание догматизма в научных исследованиях, заведшими изотопную геохимию в тупик. Практика показывает, что В.Б. Поляков не понимает существа проблемы. В этой работе он разбирает два вопроса, хотя их отдельно не подчёркивает:
1) – конструирование, или лучше уточнение условий теоретического вывода двух ГТ – кальцит - магнетитового и кальцит – графитового (решение прямой задачи). При этом он использует более совершенные с его точки зрения приёмы, хотя при этом не доказывает, почему используемые им приёмы дают более совершенный результат, а также вообще, зачем нужны эти дорогостоящие работы по «совершенствованию геотермометров», если за этим ничего больше не стоит. К тому же он использовал сомнительный метод выражения уравнения ГТ - полиномиальные уравнения высоких степеней, которые ещё более затемнили смысл ИГТ, поскольку не получена интерпретация коэффициентов при x= 106/T2; недостаток, типичный для ранних версий ИГТ более простых модификаций. Тем более, что приём этот не новый. Он применялся геохимиками, например, J.Kawabe (1978), Y. Bottinga [38] и др., ещё в те самые 60- 70 годы.
2) - применение этих ИГТ для определения температуры образования карбонатитов (решение обратной задачи). Здесь он не только не сказал ничего нового в методике решения задачи, но и догматически повторил затасканные приёмы тех самых «60-х и 70-х годов», совершив при этом те же ошибки:
- использовал только единичную пару минералов;
- сама термопара составлена из минералов, взятых с «потолка»;
- не доказал наличие между этими минералами изотопного равновесия;
- наконец, взяв для контроля вторую пару, по умолчанию предположил наличие между тремя минералами одновременного изотопного равновесия, что существующая теория изотопного обмена не рассматривает. Эти методические ошибки перечёркивают все старания В.Б. Полякова и делают их бесполезными.
3. Механизм выделения минералов.
Под механизмом выделения минералов понимается химическая реакция, ведущая к выделению минерала [23]. Решение этой проблемы способствует выяснению условий образования минералов, а, следовательно, и вмещающих минерал горной породы. Задача о механизме выделения минерала является одной из обратных задач геологии, направленных на установление условий образования горных пород (руд).
Таблица 1. Примеры механизмов выделения гранатов.
|
№№ п.п. |
Реакции |
Авторы |
|
1 |
An + 4Opx + Spin →Grt |
McLelland J.M.,1980 |
|
2 |
2An + 4Opx + FeO → Grt |
McLelland J.M.,1980 |
|
3 |
Px + Spin → Grt + Ol |
Leeman W.P., 1970 |
|
4 |
Cor + 4Hyp→ 2Grt + 3Qw |
Hensen B.J., 1971 |
|
5 |
Sil + Hyp → Grt + Qw |
Hensen B.J., 1971 |
|
6 |
Bio + Cor → Grt + Sil + Qw |
Thompson A.B.,1976 |
|
7 |
2Bio + 7Cor → 10Grt + 6Mus + 3Qw |
Thompson A.B.,1976 |
|
8 |
Hyp + Kf + W → Grt + Bio + Qw |
Ellis D.J., 1980 |
|
9 |
Ol + Spin + Qw → Grt + Cor |
Hensen B.J., 1972 |
|
10 |
2An + 2Ol + 2Opx → Grt + Cpx |
McLelland J.M.,1980 |
|
11 |
8Stav + 12Qw →4Grt + Chl + 30Kya |
Федькин В.В., 1975 |
|
An - анортит, Chl - хлорит, Corr- корунд, Dol- доломит, Fo - форстерит, Grt- гранат, Hyp- гиперстен, Kya- кианит, Ol – оливин, Phl – флогопит, Px - пироксен, Qw – кварц, Sil - силиманит, Spin - шпинель, Stav – ставролит, W - вода , Wol - волластонит. |
||
Попытки решения этой задачи привели к появлению громадного количества надуманных химических реакций, по мнению авторов, описывающих особенности процесса природного образования минералов. В качестве примера в табл. 1 и 2 даны примеры механизмов образования некоторых минералов (на примере граната). Из них видно, что один и тот же набор минералов может быть получен при различных исходных наборах минералов, и наоборот, одинаковые исходные комплексы минералов могут давать различные конечные члены реакций. Части реакций с одинаковыми комплексами минералов выделены жирным шрифтом. Некоторые реакции при одном и том же наборе минералов в обеих частях реакций отличаются только коэффициентами при этих минералах, которые в дальнейшей работе совершенно не учитываются.
Таблица 2. Примеры механизмов образования породообразующих минералов.
|
№№ п.п. |
Уравнения |
Авторы |
|
1 |
4Bio + 21Qw +5Cor = 14Grt+ 12Kf +12H2O Bio + 52Qw+ 5Sil = 8Grt +9Kf + 2H2O |
Thompson A.B.,1976 |
|
2 |
Bio + Sil + 2Qw = Grt + Kf + H2O 4Bio +3Cor + 3Qw = 6Grt + 4Kf + 4H2O |
Hensen B.J.,1971 |
|
4 |
4Cor + 3Mus = Bio + 10Sil + 11Qw 8Grt + 9Mus = 3Bio + 14Sil + 13Qw |
Thompson A.B.,1976 |
|
7 |
2Bio + 6Sil + 9Qw= 3Cor+ 2Kf + H2O Bio + Sil + 2Qw = Grt + Kf + H2O |
Hensen B.J.,1971 |
|
8 |
4Bio + 8Sil + 19Qw = 5Cor + 4Kf + 4H2O 4Bio + 6Sil + 9Qw = 5Cor + 5K2O + 4H2O |
Zeck H.P.,1970 |
Подобная ситуация сложилась и в рудной геологии. При таком разнообразии реакций выделения минералов не ясно, какая же реакция описывает реальный процесс минералообразования? Все имеющиеся уравнения выделения минералов представляют собой либо продукт произвольного толкования результатов экспериментальных исследований, либо вообще «сочинением на вольную тему». В обоих случаях наблюдается произвол в составлении реакций образования минералов. Например, В.И. Лучицкий [29], описывая замещение роговой обманки (далее Amp), приводит реакцию 5Amp + 7W ⇒ 2Ep + Chl + Act + Qw + 2H4Mg2Al2SiO9 (Act – актинолит) и пишет: «Обыкновенно одновременно развивается эпидот Ep (более высокотемпературный) и хлорит Chl (более низкотемпературный)…». Но если минералы в окрестности одной точки появляются при разных температурах, значит, они не одновременны. Следовательно, данная реакция должна быть разбита минимум на две самостоятельные реакции [23]. Подобный подход иллюстрирует и реакция 11 (табл.1), поскольку гранат и хлорит образуются при разных температурах. Особенно же одиозны реакции с участием флюидных компонентов. В целом, все эти реакции не отражают реальные условия образования минералов в природе.
4.Задача об источниках вещества.
Задача об источниках вещества - одна из фундаментальных задач геологии. Её решение позволяет осветить не только проблему поисков месторождений, но и способствует решению важных в научном отношении задач, связанных с вопросами образования горных пород и руд. Принципиальным отличием этой проблемы от предыдущих задач является то, что, если во втором случае имеются теоретические положения, сравнение с которыми позволяет оценить качество их использования, то в данном случае полностью отсутствуют какие-либо теоретические положения. Существующие критерии решения этой проблемы базируются на интуитивных положениях, теоретически не обоснованных. Это существенно затрудняет анализ качества решения задачи об источниках вещества.
В основе анализа лежит механическое сравнение либо набора элементов, либо их количественных характеристик, либо отношений концентраций элементов в изучаемых объектах с таковыми, принятыми за эталон. Например, присутствие в породах Be, Sn, W и других элементов, по мнению ряда авторов, говорит об участии гранитоидного вещества. Наличие Ni, Co, Cu и т.д. позволяло говорить об участии вещества основных и ультраосновных пород и т.д. Здесь даже появился специальный термин- геохимическая специализация. Заметим, что применяемые методы содержит методическую ошибку: здесь использован метод аналогий (эталонов), но метод аналогий является только методом построения гипотезы [1], а не является методом доказательства.
Проблема источников любого, в том числе и рудного вещества раскладывается на две составные части [27]: «Определение глубины нахождения источников рудного вещества» и «Определение состава источника вещества». Эти проблемы отвечают на вопросы: где и что находится в источнике вещества? В геологической практике обычно решается преимущественно вторая качественная задача, в то время как первая задача обходится стороной.
При анализе этой проблемы необходимо учитывать разделение задачи на этапы: прямая и обратная задачи. Стандартное соотношение между ними приведено на рис.2. В случае решения прямой задачи имеется объект исследования, природа которого достоверно известна; на объекте выделяется некоторый минерал, изучается его состав (например, изотопы С, О, S или концентрации других элементов) и считается (обратная
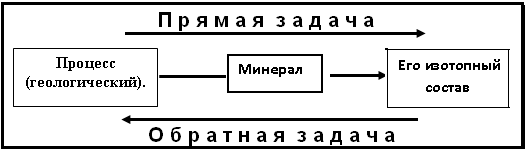
Рис. 2.Практическое взаимоотношение между прямой и обратной задачами
задача), что этот состав характеризует описываемый геологический процесс. Этот способ интерпретации широко используется в рудной геологии, металлогении, гидрогеологии и пр. На самом деле здесь есть две серьезные ошибки. Первая ошибка состоит в том, что игнорируется зависимость состава минерала от физико-химических условий (ФХУ) минералообразования. С учетом этого решение прямой задачи схематически отражается рис. 3. Вторая ошибка состоит в следующем. При решении обратной задачи изотопный состав минерала характеризует некоторые ФХУ его образования. Но одни и те же условия могут характеризовать различные процессы, выбор которых должен быть доказан, но это не делается.
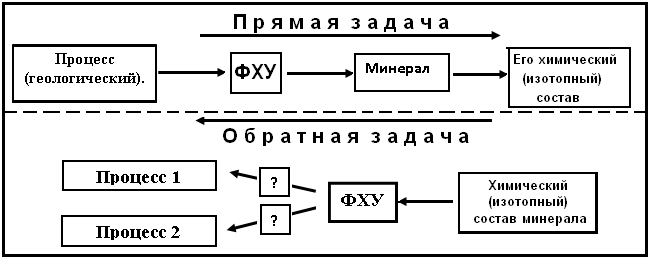
Рис.3. Истинное взаимоотношение между прямой и обратной задачами.
Можно говорить о двух направлениях решения этой задачи:
1. Использование элементов;
1а). Концентраций элементов;
1б). Отношений концентраций элементов;
2). Использование изотопов элементов.
1а). Использование концентраций элементов для оценки источников вещества является наиболее древним методом. Пример такого подхода приведен выше. Прежде всего, он основан на геохимической специализации магматических образований, т.е. на том, что какие-то элементы встречаются только в одних породах, другая часть - в других породах и отсутствуют в породах первого типа, и поэтому присутствие этих элементов в образце позволяет предположить характер источника этих элементов. Эта интерпретация всегда интуитивная, и потому достаточно не надёжная.
1б). Считается, что отношения элементов является более надёжными. Эта методика также основана только на интуитивных представлениях. Особенность её в том, что, как показывает теория (термодинамика, петрохимия), отношения концентраций элементов являются индикаторами условий образования минералов, т.е. эти отношения чутко влияют на колебания T, P и других термодинамических параметров. На этом даже основан метод определения условий образования минералов (см. выше - баро- и геотермометры). Поэтому для решения задачи об источниках элементов необходимо снять влияние этих параметров. Но на практике, это никогда не делается.
Наиболее одиозны решения подобных задач с применением изотопов. Изначально проблема определения источников вещества решалась также как и при анализе элементного состава пород и минералов. Выбирались эталоны с уже известным изотопным составом слагающих их компонентов и по степени близости измеренных значения изотопных отношений изучаемого минерала делались выводы о степени участия вещества эталонов. Например, по степени близости величины δ34S к метеоритному стандарту судят о доли участия метеоритного вещества; по степени близости величины δ18O определялась доля метеорных вод в объекте и пр. Подобных ляпов в изотопной геохимии множество [31, 32, 33]. Они ближе всего к этюдам жонглирования. Определялись даже доли Х участия в измеряемом объекте вещества эталона. Подобный подход обладает рядом существенных недостатков:
1) сама формула для расчёта величины Х – это грубое приближение; ее анализ показывает, что она отражает не доли смешивающихся вод, а доли растворенных в этой воде веществ.
2) Природа эталона не доказывается, а принимается априорно, т.е. произвольно, субъективно.
3) Изотопный состав элементов эталона также не доказывается, а принимается из геологических соображений, т.е. также произвольно.
4) Не учитывается, а значит, не вносятся поправки, учитывающие влияние факторов физико-химического характера (T, минерализация воды, диффузия компонентов и др.); дело в том, что многие особенности распределения изотопов могут быть объяснены влиянием этих факторов.
5) Грубой ошибкой является отсутствие контроля (проверки) достоверности получаемых выводов.
Особенность задачи состоит в том, что метеорные, магматические, гидротермальные потоки и флюиды – тепловые системы, поэтому к ним применимы способы описания тепловых систем, позволяя тем самым проверить [30] достоверность получаемых выводов для гидротермальных систем, например, по оловорудным системам [34]. Пренебрегая в первом приближении тепловыми потерями, запишем уравнение теплового баланса:
Q1 + Q2 = C1m1 (T01- T11) + C2m2 (T02- T12) = 0.
Здесь, Q1– количество тепла, приобретенного потоком метеорных вод массой m1 и удельной теплоемкостью С1 при нагреве от Т01 до Т11; Q2 – количество тепла, отданного потоком магматических вод удельной теплоемкостью С2 и массой m2 при нагреве от Т02 до Т12. При T11= T12 = Тгом кварца (касситерита), C1= C2, Т01= 25оС и зная отношения масс смешивающихся вод в водном потоке, можно рассчитать исходную Т02 магматических вод, гипотетические составы магматической воды и гранита, породившего этот поток, и сравнить их с уже известными параметрами этих объектов. Результаты расчетов приведены в таблице 3. Видно, что кроме неправдоподобно высоких Т магматических вод
Таблица 3.Исходные параметры магматических вод.
|
№№ п.п. |
Месторо-ждение |
Вмещающие породы (п.) |
Минер.-гео-хим. тип |
ТоС |
δ18О%о |
||
|
Т11 |
Т02 |
кварц |
вода |
||||
|
1 |
Прииска-тель |
Пегматит |
Грейзен. |
315 |
374 |
9,50 |
7,50 |
|
2 |
Лиственное |
Гранит, оса-дочные п. |
Хлорито-вый. |
350 |
1275 |
5,90 |
11,10 |
|
3 |
Невское |
Песчано-сланцевые пор. |
Пирофил-литовый. |
380 |
747 |
6,10 |
10,90 |
|
4 |
Трудненьское |
Покровы, жерла вулкана |
Многосу-льфидное. |
300 |
373 |
9,50 |
7,50 |
наблюдается несоответствие в геологическом распределении их источников: так на месторождении Приискатель с типично пространственной связью с гранитоидами, Тобр которых не ниже 500– 700оС, Т магматических вод еле-еле достигает критических значений. Для месторождений (Лиственное, Охотничье, Айнаветка) с близповерхностными условиями формирования и практическим отсутствием видимой связи с гранитоидами, Т магматических вод превышает 1000оС, типичных для свободных газов активных вулканов. Такая же противоречивость наблюдается и в графах 5– 7. Величина δ18О (Qw) только в двух случаях соответствует принятому изотопному составу гранитоидов, причем в тех месторождения, для которых связь с этими породами минимальна. Остальные же значения близки породам основного состава. Приняв средний изотопный состав гранитоидов δ18О= 8,5%о, оценена величина δ8О воды, изотопно равновесной гранитоидам, при полученных температурах очага. И здесь выявилось отличие от принятого для расчета смешения значения магматических вод. Таким образом, проверка не подтвердила обоснованность и показала надуманность выводов о природе гидротермальных вод потоков, сформировавших оловорудные месторождения.
В работе [30] детально рассмотрены недостатки изотопных методов. В результате подобные «эксцессы» входят в противоречие либо с геологическими данным (Г.С. Рипп, 1979), либо друг с другом (Л.К. Гуцало,1980 и др.).
5.Выход из этого положения.
5а .Теоретическая геохронология (ТГХ)[25].
При построении ТГХ использованы следующие аксиомы.
а).Определение возраста опирается на явление радиоактивного распада изотопов урана.
б). Свинец - смесь радиогенного iРbр и примесного iРbo компонентов (мы отказались от таких бессодержательных понятий как обыкновенный, первозданный, первичный и пр. свинцы). По природе Рbo – это Рb, отторгнутый вместе со своим U и перенесенный в изучаемую точку. Тогда отношение γ = 207Рb/206Рb отражает возраст этого свинца.
Смешение этих компонентов в точке А оценивается по равенству
iМрА = (iCА - iCo)/(iCp - iCo) = (1- iМоА). (3)
в). Рудный минерал содержит только радиоактивные и радиогенные компоненты: UO2 +ThO2 + Pb=100%.
Тогда распределения свинцов описывается уравнениями
iС = bi/Pb + iСо1; например, iС = ai 204C + iСр1.
В этих уравнениях iСо1 и iСр1 – соответственно составы примесной и радиогенной компонент. Здесь iС – относительная концентрация изотопа свинца iPb (i = 204, 206, 207, 208).
Мы ввели в практику исследований представления об «уравнениях компенсации» [35], что позволило расширить материал через методику определения источников вещества. Тогда для источника второго уровня (выше приведены уравнения для источников первого уровня, памятуя, что измеренные концентрации Pb отражают его состав в источнике уровня 0) уравнения компенсации имеют вид:
iСр = -(l4)ai + li;
iСо1 = kibi + Ki,
где li = Ki = iCo2 – относительные концентрации изотопов свинца; ki = -(1/Pbo2), здесь Pbo2 – валовое количество свинца в источнике уровня 2. Дополнительные детали рассмотрены в приводимой литературе.
5б . Геотермометры.
5б1. Изотопные геотермометры.
Данная В.Б. Поляковым [25] классификация ИГТ основана на второстепенных признаках. В основу вывода ИГТ нами применительно к соединениям с изотопами двух элементов (кальцит КЛ, мусковит МУ и др.) положены аксиомы [14]:
-в отобранных пробах анализируется один и тот же минерал М;
-минералы каждой пробы образованы в одно и то же время t; эти минералы образованы при одной и той же температуре Т;
-минералы находятся в термодинамическом изотопном равновесии (ТИР) с одним и тем же соединением С;
-наличия информации о составе соединения М2, равновесного исследуемому минералу М1;
-для минералов в условиях изотермичности выполняется равенство α= const для любых точках, где α = αт- показатель термодинамического фракционирования между М и С, зависимый от Т. Тогда имеет место равенство
S= (lnα2)/(lnα1)= f2(T)/f1(T) = F(T), (4)
Если на некотором интервале исследуемый минерал М1 образован при Т1, а минерал М2- при Т2, то пробы распределяются по прямой с угловым коэффициентом
S = (lnαy)/(lnαx)= fy(Ty)/fx(Tx), (5)
где Х и У- системы координат, по которым откладываются значения изотопных составов. В связи с этим всё многообразие ИГТ объединяется в группы:
А.Мономинеральные ИГТ.
Эти ИГТ основаны на анализе распределений изотопов разных элементов в одном и том же минерале (карбонаты С, О; слюды O, H; сульфаты S, O) по уравнению (1).
Б.Двуминеральные изотопные геотермометры.
Б-1.Моноэлементные ИГТ с одновременно образованными минералами. Это единственная в настоящее время форма ИГТ, применяемая при изотопных исследованиях. Теория его хорошо разработана, поэтому описание ИГТ не приводится. Она основана на реакции изотопного обмена вида M1* + M2= M1 + M2*. Следствием этого обмена является равенство (4), где Х-изотоп общего
δXm1 - δXm2 = F(T), (6)
для минералов элемента. Это выражение легко выводится из уравнения (5) при условии, что S=1 и C1= C2. Этот тип ИГТ не предназначен для решения температурных задач. Он имеет только феноменологический характер; на его основе созданы другие, описанные ниже ИГТ.
Б-2.Моно- и биэлементные ИГТ с минералами в произвольных временных отношениях. Это разновременно образованные соединения с изотопами как общих, так и различных элементов (например, окисел и сульфид), причем Тобр одного из них (эталона) и состав соединения С для него заранее известны. Для этих минералов выполняется соотношение (4). Имеются разновидности.
Моноэлементные ИГТ. Силикаты магматитов и метаморфитов образуют пары минералов с изотопами общего элемента, например, кислорода.
Биэлементные ИГТ. Пары минералов с изотопами разных элементов.
В работе [14] приведены результаты определения Тобр минералов. Такой подход позволил оценить и влияние диффузии на оценку Тобр минералов [36].
5б2. Геохимические геотермометры.
Геохимические геотермометры (ГГТ) полностью аналогичны ИГТ. Поскольку у ГГТ были обнаружены такие же ошибки, то для работы с ними были использованы те же приёмы, которые применялись для ИГТ. Анализ выражений для ГГТ показывает, что наиболее правильным является уже имеющее смысл уравнения выражение
ln(Kp/Кро) = -A/T + B, (6)
где Ко некоторое начальное значение коэффициента распределения для тех же элементов. Величина К/Ко является тогда безразмерной и допускает операцию «логарифмирования». Подобная трактовка предполагает и иное понимание членов уравнения (5), приобретающего вид lnKp/Кро) = (-ΔHо2/T2) - (-ΔHо1/T1). Подобная трактовка следует также и из [11, стр.127 - 128].
Главный вывод состоит в том, что все геотермометры- и геохимический, и изотопный - не предназначены для определения Тобр минералов не только потому, что нет доказательств термодинамических равновесий между природными минералами, но и потому, что не определим состав соединения, равновесного исследуемому минералу.
5в(3). Механизм выделения минералов.
Таким образом термодинамические геотермометры не предназначены для решения сугубо температурных задач. Выход из температурного тупика заключается в том, чтобы использовать эти геотермометры (ИГТ и ГГТ) для решения другой, не менее важной для геологов задачи- определение механизма выделения минералов в природных условиях. Методика решения этой задачи изложена в [23]. Решение задачи изначально разработано для случая использования изотопов элементов породообразующих минералов, но возможно и при использовании содержаний самих элементов. Это решение опирается на следующие аксиомы:
1).Мы абсолютизируем точку зрения Э.М. Галимова следующим образом: в реакциях природного минералообразования, в т. ч. низкотемпературного и биогенного, всегда сохраняется состояние ТИР между исследуемым минералом M и некоторым соединением C. Кажущееся нарушение этого равновесия вызывается: а) влиянием наложенных процессов (диффузия и пр.); б) ошибкой выбора компонента C (очень часто, например, при определении Тобр минералов)ты; в) методическими ошибками.
2). Термодинамическая система M- C образуется в результате распада исходного материнского вещества. ТИР устанавливается в процессе проведения химической реакции, в результате которой происходит распад одного вещества и образование другого. Об этом механизме свидетельствует формирование, например, изотопного равновесия в опытах с графитом (Банникова и др., 1987), шеелитом [26] и т.д.
Решение основано на сравнении данных термодинамических геотермометров (ГТ) с данными других ГТ, прежде всего с результатами анализов ГЖВ и/или расплавных включений (РВ). В целом решение задачи разбивается на два этапа:
1) определении состава соединений, равновесных исследуемому минералу;
2) на этой основе определение состава материнского соединения. Процедура решения задачи первого этапа исходит из анализа изотермы распределения изотопов элементов в одном или двух минералах. Ниже приведены примеры решений этих задач.
Было установлено, что в ~85% случаев выделение низкотемпературных кальцитов, как и в лабораторных условиях, осуществляется по реакции Ca+2 +2HCO3-1 = CaCO3 +CO2 + H2O. Это равновесие нарушается влиянием диффузионных процессов, прежде всего для CO2 и H2O.
Сульфиды в большинстве случаев изотопно равновесны ионам S-2, SO4-2, HSO4-1, H2S. Формирование сульфидов с участием иона S-2 возможно по гипотетической реакции вида ...→… + МеS (или МеS2) + S-2 + … . Более приемлемой оказалась реакция Pb(OH)4 + 2H2S → PbS + S-2 + 4H2O в соответствии с гипотезой И.Г. Ганеева (1977).
В кислых гранитоидах и метаморфитах установлена последовательность выделения минералов (Qw, Bio) > (Mt, Il) >Al1> Mus > Al2 > Grt(?) (Al- альбит); все минералы, кроме Mt, Il, выделились в равновесии и водой. Этому, например, близка реакция Al(OH)-4 + 3H4SiO4 + Na+ (К+) → Na(К)AlSi3O8 + 8H2O (по В.И. Рыженко и др., 1981; В.А. Алексееву и др., 1989). С учётом взглядов И.Г. Ганеева эта реакция приобретает вид Al+3 + 3Si(OH)6-2 + Na+(К+) + 2Н+1 → Na(К)AlSi3O8 + 8H2O + 2OH-1, поскольку только здесь допустимо изотопное равновесие не только по кислороду, но и водороду; ильмениты магнетиты и обычно образуются при разложении ульвошпинелей или ильменитов.
Минералы основных и ультраосновных пород- Ol, Px и др- выделяются в равновесии с СО2. Таким образом, поставщиком 18О в минералы является CO2, согласуясь с хорошей растворимостью СО2 в ультраосновных расплавах высоких давлений. Возможный эффект описывается, может быть, уравнением 2Mg+2 + Si(CO3)4-4 → Mg2SiO4+ 4CO2. Именно это обстоятельство объясняет присутствие углекислоты в высокотемпературных РВ. В этом случае роль СО2 аналогична роли воды в расплавах кислого состава.
5в(4). Задача об источниках вещества.
5в1а. Состав вещества в источниках. Компенсационные уравнения.
Эта задача решается с помощью представлений о компенсационных уравнениях (или уравнениях компенсации). Эти понятия заимствованы из теории диффузии. Полное описание методики приведено в работе [35]. Здесь выделяются две составляющие: А. математическая сторона вопроса; Б. сущность полученных выражений.
А. Уравнения вида Y = AX + B характеризуются парой чисел (А, В). Используя последние как некоторые координаты, получаем прямую вида B = аA + в. Если это уравнение выполняется, то такая совокупность прямых называется пучком прямых, пересекающихся в одной точке (точке кроссовера) с координатами (Ао, Во), определяемых как а = -Ао и в = Bо. Уравнение вида B = (-Aо)A + Bо называется компенсационным уравнением.
Б. Уравнение Y = AX + B описывает распределение компонентов, характеризующих состав вещества в источнике уровня 0 и образующихся при смешении исходных компонентов как минимум из двух источников более глубокого уровня 1. Применение к ним представлений о «компенсации» приводит к выводу о том, что параметры (Ао, Во) как раз и описывают состав одного из исходных компонентов в источнике уровня 1, а множество выборок из источников уровня 0, образованных из вещества уровня 1, образуют семейство этих выборок. В полном виде теория разработана для изотопов Pb (см. выше), так как в этом случае априорно известен один из источников вещества- радиоактивный распад.
Приведём несколько примеров [35].
1) Кислые изверженные и метаморфические породы. Распределения Fe2 и Mg2 в биотитах описываются уравнениями вида Mg2 = АFe2 + В; уравнение компенсации иллюстрируется на рис.3. Согласно ему в источнике уровня 1 концентрации элементов в биотитах составляли: Fe21= 2,17 и Mg21 = 2,42 кристаллохимических единиц.
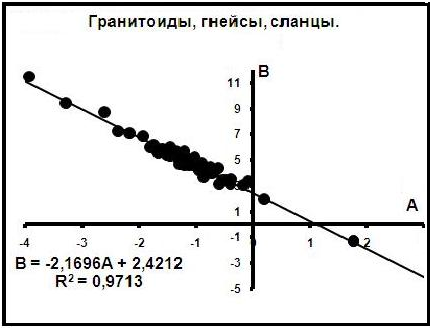
Рис.3. Диаграмма компенсации в биотитах.
2). В нефтях изучены распределения физических свойств: плотность d420 и показатель преломления nD20, определённый при 20оС для
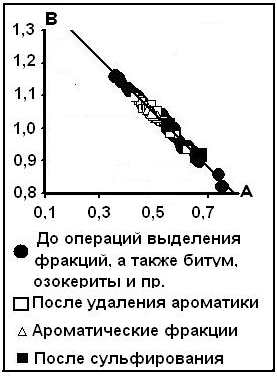
Рис.4. компенсационная диаграмма физических свойств нефти.
спектральной линии D натрия; d420 –плотность, определённая при 20оС и приведённая к плотности воды с Т = 4оС. Общее выражение этой связи дано ещё В.П. Исаевым (1972) в виде уравнения nD20 = 2,0137d420 – 2,112. Анализ распределения этих параметров по конкретным месторождениям показал сохранение подобной зависимости на всех изученных объектах; она имеет обобщённый вид nD20 = Ad420 +B. Это позволяет для нефтей построить компенсационную зависимость, данную на рис.4, из которого видно, что все нефти имеют один и тот же источник уровня 1, в котором физические свойства нефти описываются значениями do= 0,827 г/см3и no = 1,459.
3) Изучение изотопного состава кальцитов различных карбонатитов показало, что и здесь достаточно хорошо выполняются уравнения вида δ13C = Aδ18O + B. Построенная по этим данным компенсационная диаграмма имеет вид, представленный на рис.5. Эти данные показывают,
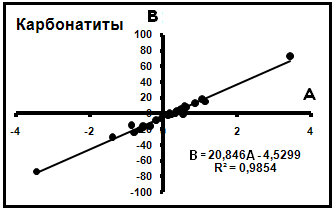
Рис.5. Компенсационная диаграмма кальцитов карбонатитов.
что карбонатиты также имеют один общий источник (по составу), для которого установлены значения δ13C = -4,53 и δ18O = 20,845%о.
5в1б. Состав вещества в источниках. Бикомпенсация.
Если выделяются несколько семейств, в общем-то, независимых друг от друга по условиям образования, то каждое семейство характеризуется парой чисел (g,G), играющих роль координаты точки в некотором фазовом пространстве и позволяющих объединить их через линейное уравнение первого порядка. Эти уравнения, в случае своего существования, называются уравнениями бикомпенсации, а множество прямых линий, обобщаемых этим уравнением, образуют надсемейство. Параметры уравнений бикомпенсации характеризует свойство источника вещества уровня 3. Такие случаи установлены при анализе угля, нефтей, карбонатов. Пример подобной ситуации отражён на рис.6 для кальцитов разной природы. Выделяются два надсемейства.
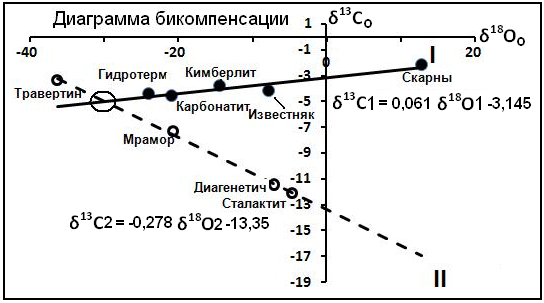
Рис.6. Диаграмма бикомпенсации по карбонатным семействам.
Возможный родовой источник описывается координатами точки пересечения (на рис.6 обведено кружком) диаграмм надсемейств: δ18Oоо ≈ -30%o (SMOW) и δ13 Cоо ≈ -5%о (PDB), что позволяют оценить источники изучаемых элементов. Изотопный состав углерода не информативен. Интересно наличие лёгкого кислорода (δ18Oоо ≈ -30%o (SMOW)), отсутствующего на Земле. В некоторых метеоритах (Лаврухина, 1992) кислород имеет значение δ18O = -65‰. Близкие величины обнаружены в тонкозернистых межпланетных частицах. Анализ образцов космического аппарата "Генезис" (Genesis) показал, что на Солнце концентрация 16О (отношение количества 16О к общему количеству кислорода) существенно выше, чем на Земле [37]. Таким образом, изначально кислород земных карбонатов, возможно, имел космогенную природу.
5в2. Глубина нахождения источника.
Одна из важнейших и наименее решённая обратная задача геологии. Значительную роль она играет в поисковых методах. В российской геологии некоторые разделы этой проблемы рассматривались А.П. Солововым, предложившим эмпирические методы её решения по данным геохимических (металлометрических) поисков; значительную роль сыграли работы В.М. Дубова, пытавшего решать эту задачу с использование параболических уравнений. Имеются другие попытки решения проблемы, но сейчас все они имеют только историческое значение. Ниже кратко рассмотрен подход к решению этой задачи применительно к гидротермальному процессу согласно работе [27]. Это решение имеет наиболее общий характер, предназначено не для непосредственного использования, а как основа дальнейшего анализа проблемы.
При разгрузке гидротермального потока (раствора) происходит выделение и осаждение соединений (минералов, газов и пр.). В первом приближении этот процесс можно выразить уравнением (7), в котором А, В- реагирующие компоненты гидротермального потока, АВ- продукты
А+ В = АВ + Qr (7)
реакции, Qr - тепловой эффект реакции при Р= const. Согласно представлениям физической химии Q = - ΔHr, где ΔHr- энтальпия реакции.
Гидротермальный поток рассматривается как объект, обладающий тепловой энергией Qт, поэтому всегда предполагается, что Qr = Qт. В таком виде это равенство не точно, поскольку не учитывает кинетическую Qк, потенциальную Qп энергии и потери А энергии при движении потока. С учетом этих факторов уравнение осаждения преобразуется в уравнение:
Qr+ Qт + Qк+ Qп + А= 0.
Подставляя соответствующие выражения для названных видов энергий, учитывая, что Qт = mMDHr и mM – масса выделившегося вещества, и полагая в грубом приближении А = 0, получаем феноменологическое уравнение (8)
Срmр(Тк- Тн) + mрgh + mрvр2/2 + mMΔHr = 0 (8)
решения задачи о глубине нахождения источников вещества, в котором Ср и mр- соответственно теплоемкость и масса потока, Тк и Тн – температура начальная и конечная (соответственно зарождения и разгрузки) потока, h – глубина нахождения (зарождения потока) источника; vр – скорость движения потока (раствора). В этом уравнении неизвестными являются Тн, h, t (время движения потока), известными- Qт, Ср, mM,Тк, mр.
Рассмотренные общие принципы решения задачи можно применять не только для изотопов, но и для элементов, которые, так или иначе, разделяются при движении и разгрузке гидротермального потока. Тогда, видоизменив уравнение (6), можно записать его в виде реакций обмена для трёх элементов
AX + Y = AY + X + Q1, (7а)
AX + Z = AZ + X + Q2, (7б)
BX + Y = BY + X + Q3. (7в)
Анализ уравнений (7а)- (7в) показывает зависимость величины Qi от вида компонентов системы. Это означает, что решение задачи зависит от составляющих этих уравнений, т.е. от формы нахождения вещества в потоке и минерале. Объединяя уравнения (7а)- (7в) с уравнением (8), получаем систему уравнений (9) для решения задачи на конкретном месторождении
m2С2(T2- T02)+ m2g(h2 + Δh2) + m2v22/2 = M2 ΔHp2 (9б)
m3С3(T3- T03)+ m3g(h2 + Δh3) + m3v32/2 = M3 ΔHp3. (9в)
по трем элементам, где Δhi – высотные отметки точек наблюдения от некоторого исходного уровня. Решение системы упрощается, если: T01 = T02= T03, - в источнике равные температуры для всех элементов; v1= v2= v3,- скорости движения потока одинаковые для всех элементов. Если
Δh1 ≠ Δh2 ≠ Δh3, то решение задачи опирается на несовпадение в пространстве максимумов накопления элементов, или другими словами – на анализ геохимической зональности элементов на месторождении.
Для решения задачи наиболее удобными являются распределения изотопов в одном минерале, например, О и С в кальците, S и Pb – в галените; или наличие в одном минерале нескольких элементов- спутников, поскольку для них можно полагать одну и ту же глубину нахождения источника, близость путей их миграции, ослабленное влияние внешней среды на эту миграцию.
Таким образом, решение задачи о глубине нахождения источника (рудного) вещества является комплексной и предполагает разработку следующих частных проблем:
1.Установление и анализ геохимической зональности, (ореольно- геохимическая часть программы).
2.Установление форм нахождения элементов в областях наибольшей концентрации элементов (минералогическая часть программы)
3.Анализ распределения тепла в процессе перемещения и разгрузки гидротермального раствора (теплофизическая часть программы, наименее разработанная часть программы).
4.Определение механизма минералообразования, влияющего на велличину ΔH, и обоснование вида уравнения (1) (физико-химическая часть программы).
6.Заключение
Таким образом, здесь проведён анализ небольшого количества приёмов, использованных при проведении реальных геологических исследований. В стороне осталась не только научная интерпретация большого количества других материалов, но и такие явления, как разбазаривание финансовых средств, отпускаемых на проведение этих работ, несмотря на то, что геологи постоянно ноют по поводу их недостатка.
Эти доказательства получены ещё в 80-х годах прошлого века. Тем не менее, эти выводы совершенно не учитываются, даже нет попыток их опровержения. Последнее напоминает бойкот выявленных закономерностей. Учёные геологи продолжают барахтаться в гнилом болоте геологического примитивизма, поскольку это намного легче, безопаснее и экономически, и карьеристски выгоднее.
Литература
1.Макаров В.П. Некоторые вопросы методологии научного геологического познания./ Материалы X научного семинара «Система планета Земля». М.: РОО «Гармония строения Земли и планет». 2002, С.19- 26. URL: http://www.lithology.ru/node/524.
2.Ленин В.И. Материализм и эмпириокритицизм. М.: Издательство политической литературы, 1984. 107 с.
3.Энгельс Ф. Диалектика природы. М.: Издательство политической литературы, 1953. 348 с.
4. Макаров В.П. Некоторые методологические проблемы геохронологии. / Материалы XI научного семинара «Система планета Земля». М.: РОО «Гармония строения Земли и планет». 2003, С.71- 95.
5. Шуколюков Ю.А., Горохов И.М., Левченков О.А. Графические методы изотопной геологии. - М.: Недра. 1974. 207 с.
6. Перчук Л.Л. Теория фазового соответствия и геологическая термобарометрия.// Соросовский Образовательный журнал. Науки о земле. 1996, №6. URL: http://www.pereplet.ru/obrazovanie/stsoros/118.html
7. Макаров В. П. Методические аспекты геохимической геобарометрии./ «Геология и минеральные ресурсы Европейского Северо- Востока России: новые результаты и перспективы». Материалы XIII Геологического съезда республики Коми. - Сыктывкар, 1999, С. 60- 63.
8. Перчук Л.Л. Биотит - гранатовый геотермометр. Доклады АН СССР, 1967,177, 2, С.411-414.
Перчук Л.Л. Коррекция биотит - гранатового термометра для случая изоморфизма Mn = Mg + Fe гранате.//Докл. АН СССР, 1981, Т.256, 3. С.441-442.
9. Перчук Л.Л., Мишкин М.А., Котельников А.Р и др. Термодинамические условия метаморфизма пород Ханкайского массива./Очерки физико-химической петрологии. М.: Наука, Т.6, 1977. С.139 – 167.
10. Курепин В.А. Термодинамические условия образования гранат – кордиерит-биотитовой ассоциации в бердичевских гранитоидах (Украинский щит).//Минералогический журнал, 1991, 1. С.76 – 87.
11. Вуд Б., Фрейзер Д. Основы термодинамики для геологов. М.: Мир, 1981. 184 с.
12. Венедиктов В.М., Глевасский Е.Б., Голуб Е.Н. и др. Пироксены породообразующие Украинского щита. Киев: Наукова Думка, 1979. 228 с.
13. Магматогенная кристаллизация по данным изучения включений расплавов. Тр. Ин-та геологии и геофизики, вып. 264. Новосибирск: Наука, 1975.
14. Макаров В.П. Изотопные геотермометры./ Материалы XIII научного семинара «Система планета Земля». М.: РОО «Гармония строения Земли и планет». 2005, С.93- 115.
15. Перчук Л.Л. Равновесие породообразующих минералов. М.: Наука, 1970. 391 с.
16. Чайников В.И. // Геохимии и минералогия магматогенных образований. – Владивосток: изд. ДВГИ, 1966. С.64.
17. Перчук Л.Л., Аранович Л.Я. Усовершенствованный Bi-Gr геотермометр. Коррекция на содержание F в биотите.//Доклады АН СССР, 1984, 277, 2. С.471 – 475.
18. Ваганов В.И., Соколов С.В. Термобарометрия ультраосновных парагенезисов.- М.: Недра, 1988. 149 с.
19. Shieh Y.N., Taylor H.P. Oxygen and hydrogen isotope studies of contact metamorphism in the Santa Rosa range, Nevada and other areas.//Contrib. Miner. and Petrology, 1969, 20, 4. P.306-356.
20. Rye R.O., Schuiling R.G., Rye D.M., Jensen J.B.N. Carbon, hydrogen, and oxygen isotope studies of the regional metamorphic complex at Naxos, Greece.// Geoch. Cosmochim. Acta, 1975, 40, 9. P.1031 – 1049.
21. Campbell F.A., Ethier V.G. Sulfur isotopes, iron content of sphalerites and ore texture in the Anvile Ore Body, Canada.//Econ. Geol., 1974, 69, 4. P. 482-493.
22. Салье М.Е. и др. Фракционирование изотопов кислорода в минералах полиметаморфических комплексов докембрия. Л.: Наука, 1983, 157 с.
23. Макаров В.П. О механизме выделения минералов. / Материалы XVI научного семинара «Система Планета Земля». М.: ТОО «Гармония строения Земли и планет», 2008. С.265 – 300. URL: http://www.lithology.ru/node/861.
24. Макаров В.П. Вопросы теоретической литологии. 1. Геологическое наблюдение./Междунар. научно-практическая конференция «Перспективные инновации в науке, образовании, производстве и транспорте». Одесса: Черноморье, 2007, Т.15. С.24-31.URL: http://www.lithology.ru/node/624.
25. Макаров В.П. Основы теоретической геохронологии. / Мат-лы XII научного семинара «Система планета Земля». М.: РОО «Гармония строения Земли и планет». 2004, С.228- 253. URL: Викизнание.
26. Устинов В.И., Гриненко В.А. Изотопные равновесия в минеральных ассоциациях.// Геохимия, 1990, 3. С.307- 315.
27. Макаров В.П. Вопросы теоретической геологии. 12. Основы теории решения задачи об источниках вещества. А. Общие вопросы. Б. Выводы основных уравнений./ Международная научно-практическая конференция «Научные исследования и их практическое применение. Современное состояние и пути развития ‘2007». //Одесса: Черноморье, 2008, Т.17. С. 12-47.
Макаров В.П. Источники вещества. Вопросы теории. URL: http://www. lithology.ru/node/870.
Макаров В.П. Особенности распределений относительных содержаний изотопов свинца. //Советская геология. 1991, 6. С. 56 - 61.
Макаров В.П. О природе обыкновенного свинца в минералах. //Отечественная геология.1994. №5. С. 67 - 76.
28. Поляков В.Б. Новые калибровки углеродных и кислородных изотопных геотермометров (карбонаты, графит, магнетит). URL: http://geo.web.ru/conf/alkaline/2007/62.pdf.
29. Лучицкий В.И. Петрография. Т. 2. М-Л.: Госгеолиздат, 1949. 438 с.
30. Макаров В.П. Некоторые свойства геохимических геотермометров./ Материалы XV научного семинара «Система планета Земля». М.: ЛКИ, 2007, С.142- 159.
31. Ветштейн В.Е. Изотопы кислорода и водорода природных вод СССР. Л.: Наука, 1982. 216 с.
32. Природные изотопы гидросферы. М.:Недра, 1975. 277 с.
33. Гриненко В.А., Гриненко Л.Н. Геохимия изотопов серы. М.: Наука, 1974.271 с.
34. Борщевский Ю.А. и др. Систематика оловорудных месторождений Северо–Востока СССР по изотопно– кислородным данным.//Сов. Геология. 1982, 2. С. 94-105.
35. Макаров В.П.«Явление компенсации» - новый вид связи между геологическими объектами./Мат-лы I междун. Научно-практич. конференции «Становление современной науки-2006». Т.10. Днепропетровск: Наука и образование, 2006. С. 85-115. URL: http://lithology.ru/node/817
36.Макаров В.П. О влиянии диффузии на формирование изотопного состава подземных вод. URL: http://hydropetroleum.ru/conference/smej/sm10.pdf
37. Разбившийся Genesis и происхождение Солнечной системы. URL: http://nauka21vek.ru/archives/15052
38. Bottinga Y. Calculation of fractionation facrtors for carbon and oixygen isotopic exchange in the system calcite-carbon dioxide - water.//J. of Physical Chemistry, 1968, 72, 3, 800-808.
- Войдите или зарегистрируйтесь, чтобы получить возможность отправлять комментарии